
Varianti Istologiche
I reni sono organi capsulati che si trovano nel retroperitoneo circondati da tessuto adiposo. Il parenchima renali si compone di due zone, una corticale e una midollare.
Il rene è caratterizzato dalla possibile insorgenza di tumori benigni, tumori maligni e può essere sede di metastasi di tumori insorti in altre sedi.
Tumori benigni
I tumori benigni più frequenti sono l’oncocitoma e l’angiomiolipoma.
Oncocitoma
Gli oncocitomi rappresentano dal 5 al 9% delle neoplasie che possono insorgere nel rene. Insorge maggiormente negli uomini e, spesso risulta difficile la diagnosi differenziale preoperatoria (attraverso le indagini radiologiche) con altre neoplasie (es. carcinoma renale a cellule chiare). Si tratta solitamente di neoplasie singole e monolaterali, anche se è possibile il riscontro di masse bilaterali.
Macroscopicamente si tratta di masse ben circoscritte di colore marrone, circondate da una pseudocapsula e che al taglio presentano un’area grigiastra centrale di colore solitamente bianco-grigiastro traslucido a margini irregolari o stellati (cicatrice centrale). Possono essere emorragiche ma non dovrebbe essere presente aspetto necrotico. Al microscopio ottico si osservano cellule rotonde, ovali o poligonali con una colorazione rosa uniforme e citoplasma di aspetto finemente granulare poiché sono presenti numerosi mitocondri.
Per la diagnosi può essere necessaria la combinazione degli aspetti morfologici che l’anatomopatologo osserva al microscopio ottico con l’analisi di proteine che sono presenti nel tessuto tumorale tramite metodiche di immunoistochimica.
Angiomiolipoma
L’angiomiolipoma è una lesione benigna sporadica appartenente alla famiglia dei PEComi costituita da cellule, caratterizzate da un ritmo di crescita lento, che formano vari tessuti all’interno della massa: tessuti vascolari a parete spessa con dilatazioni caratteristiche (vasi aneurismatici), tessuto muscolare e tessuto adiposo.
Può insorgere anche nel contesto della sclerosi tuberosa familiare, malattia ereditaria, in cui si osservano lesioni multiple con possibile estensione maggiore e possibile coinvolgimento della vena renale o della vena cava.
È più frequente nelle donne di mezza età, probabilmente a causa dell’influenza ormonale. L’angiomiolipoma è una lesione neoplastica benigna con caratteristiche radiologiche peculiari che lo rendono identificabile radiologicamente nella maggior parte dei casi.
Macroscopicamente la massa è di colore giallo o grigio, a seconda della componente che predomina tra i tessuti che lo compongono.
Tumore papillare a cellule chiare
Una entità recentemente inserita nella nuova classificazione dell’Organizzazione mondiale della Sanità (WHO V classification) è il tumore papillare a cellule chiare, separato dai carcinomi a cellule chiare renali, poiché dopo una adeguata resezione chirurgica non è stata descritta nessuna metastasi e nessuna recidiva.
Rappresenta il 3 – 4% di tutti i tumori renali, sono solitamente di piccole dimensioni (media 2 cm), insorgono con la stessa presenza negli uomini e nelle donne.
Macroscopicamente si presenta, solitamente, come una massa ben circoscritta, che può presentare aree emorragiche o cistiche. La neoplasia è costituita da cellule con citoplasma di colore biancastro, che si dispongono a formare strutture digitiformi (papillari) con un asse stromale fibrovascolare. Caratteristica fondamentale di queste cellule è il basso grado nucleare: le cellule, infatti, presentano alterazioni blande se paragonate alle cellule del tessuto sano da cui originano. È possibile osservare aree emorragiche ma risulta privo di aree di necrosi.
Talvolta nell’iter diagnostico anatomopatologico possono essere necessarie ulteriori indagini per giungere alla diagnosi di tumore a cellule chiare papillare renale.
Tumori maligni del rene: carcinoma renale
Vi è poi la categoria di tumori maligni del rene, chiamati genericamente “carcinoma renale”, ma che possono presentare diversi aspetti morfologici e molecolari.
Il carcinoma renale a livello mondiale rappresenta il 2-3% delle neoplasie con un rapporto uomo/donna di 2 a 1. La maggior parte dei pazienti ha più di 50 anni.
Fattori di rischio riconosciuti sono fumo di sigaretta (probabilità doppia di sviluppare tumore del rene), obesità, ipertensione arteriosa, malattia renale cistica acquisita dovuta a malattia renale allo stadio terminale, fattori ambientali e occupazionali quali l’esposizione cronica ad alcuni metalli o ad alcune sostanze (ad esempio cadmio, asbesto, fenacetina, trielina, idrocarburi policiclici aromatici e solventi industriali).
Nella diagnosi della neoplasia l’anatomopatologo descrive l’architettura delle cellule tumorali e il loro aspetto, arrivando alla definizione del tipo istologico, che ha un suo significato biologico importante anche nella valutazione della gestione terapeutica.
Carcinoma a cellule chiare
Il carcinoma a cellule chiare è il tumore del rene più comune negli adulti ed origina dalle cellule che rivestono i tubuli renali.
Il 95% sono tumori sporadici singoli e la presenza di masse bilaterali e multicentriche è caratteristica della malattia ereditaria. La forma ereditaria è legata alla sindrome di Von Hippel Lindau con maggiori probabilità di sviluppare tumori benigni e maligni.
Macroscopicamente si presenta solitamente come una lesione che origina dalla regione corticale renale, può essere multilobata e ha colore giallo-ocra (colorazione dovuta all’accumulo nelle cellule di glicogeno e lipidi) con possibile presenza di calcificazioni, aree emorragiche o aree colliquate nei casi con necrosi tumorale. Possibile riscontrare anche aree cistiche.
All’esame microscopico la neoplasia risulta composta da cellule tumorali che, hanno come elemento distintivo il citoplasma chiaro per via dell’accumulo di acidi grassi e che crescono disponendosi a formare nidi, cordoni, tubuli o strutture pseudopapillari, microcistiche e/o macrocistiche o che possono presentare crescita solida.
Il tumore risulta riccamente vascolarizzato, da una fitta rete di capillari. Si possono osservare aree emorragiche o aree di necrosi, che, se presente, è un fattore prognostico negativo.
Per i casi dubbi l’ausilio dell’immunoistochimica può risultare utile nella diagnostica differenziale, attraverso reazioni di particolari anticorpi che legano proteine espresse dal tessuto tumorale, ‘’evidenziandole’’.
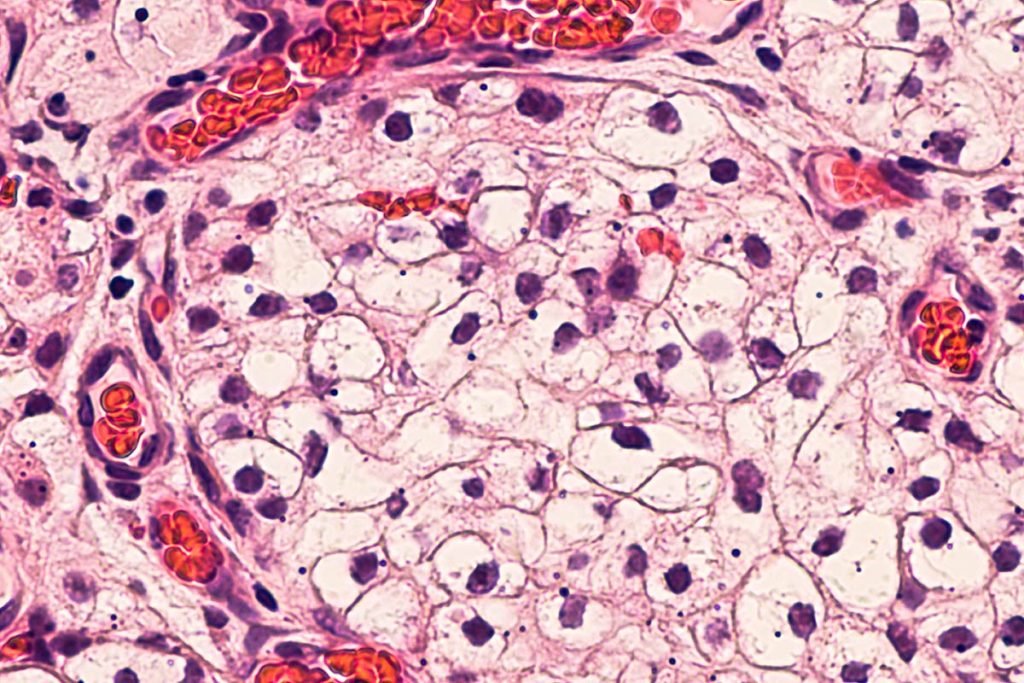
Carcinoma papillare del rene
Il carcinoma papillare è il secondo tumore più frequente del rene. Deriva, come il carcinoma a cellule chiare dall’epitelio che riveste i tubuli renali.
Insorge, nella maggior parte dei casi, dopo i 50 anni e si presenta più frequentemente negli uomini rispetto alle donne (rapporto di 1,5:1).
La maggior parte dei casi sono sporadici ma rispetto agli altri carcinomi a cellule renali può presentarsi più spesso bilateralmente o essere multifocale.
Esiste una rara forma familiare ereditaria (carcinoma papillare ereditario a cellule renali), causata da una mutazione nel gene MET, che ha la funzione di controllare la crescita cellulare.
Macroscopicamente il carcinoma papillare del rene si presenta in genere come una massa rosso-brunastra, che può presentare nel suo contesto aree grigiastre frammentabili, aree emorragiche, aree cistiche e aree colliquate se necrotico.
Al microscopio ottico questo carcinoma presenta la tipica formazione di strutture a forma di dita, definite papille, da parte delle cellule tumorali, con un asse di supporto (stromale) fibrovascolare.
Aspetti distintivi della neoplasia sono la presenza di macrofagi, la presenza di emosiderina e dei cosiddetti corpi psammomatosi (calcificazioni).
Anche per il carcinoma papillare renale per i casi dubbi l’ausilio dell’immunoistochimica può risultare utile nella diagnostica differenziale.
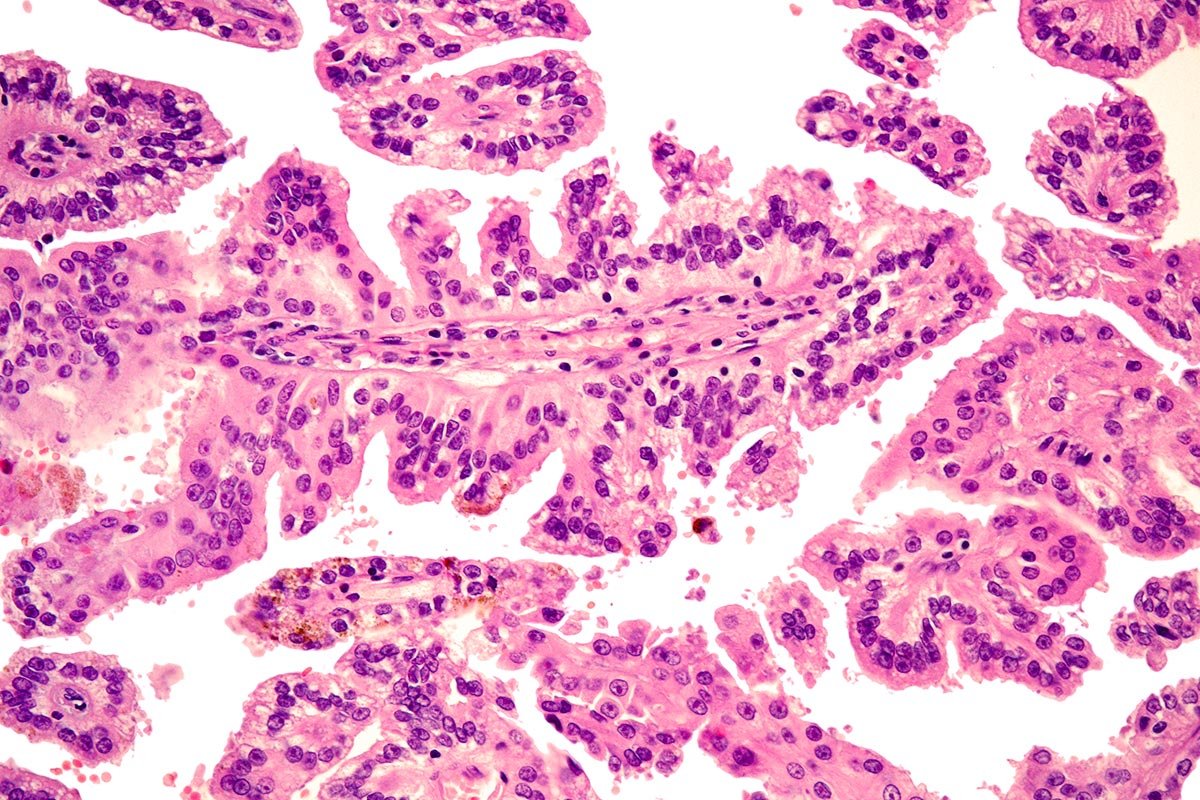
Carcinoma cromofobo
Studi dimostrano l’origine di questo carcinoma dalle cellule del nefrone distale. Nella maggior parte dei casi si tratta di carcinomi sporadici, corrisponde a circa il 5% dei campioni operatori per neoplasie renali ed è più frequente negli uomini.
Esiste una forma familiare, la sindrome di BIRt-Hogg-Dubè, che insieme predispone e aumenta la probabilità di sviluppare neoplasie tra cui quelle renali: tra esse la più frequente è lo sviluppo del carcinoma cromofobo.
Solitamente sono masse singole, di dimensioni variabili, e macroscopicamente hanno un colore marrone chiaro o beige, un aspetto pallido ed una consistenza relativamente omogenea. Spesso sono dei tumori ben demarcati, senza una vera e propria capsula.
Si possono osservare, anche se più raramente, aspetti di necrosi e emorragici, ed è possibile che presentino una cicatrice centrale biancastra traslucida (con frequenza molto inferiore rispetto a quanto avviene nell’oncocitoma renale).
Al microscopio ottico l’anatomopatologo osserva una architettura solida, o più raramente con formazione di tubuli, costituita da due popolazioni di cellule. Una popolazione cellulare con citoplasma ampio, pallido o trasparente e una popolazione con citoplasma che si colora maggiormente in rosa (simile a quello delle cellule che costituiscono l’oncocitoma) ma con caratteristiche specifiche quali ad esempio l’alone pallido o trasparente che si può osservare intorno al nucleo.
Per questa tipologia di tumori non viene applicato il sistema di grading: la maggior parte dei carcinomi cromofobi presenta spiccate atipie rispetto a quelle del tessuto non neoplastico. È perciò importante la valutazione, da parte dell’anatomopatologo, di caratteristiche come necrosi tumorale oppure la presenza di differenziazione sarcomatoide, con una parte più o meno cospicua della neoplasia che assume aspetti simili a quelli di un sarcoma, fattori che hanno un impatto negativo sulla prognosi del paziente.
Carcinoma renale con traslocazione
Nella nuova classificazione WHO è stata introdotta una categoria di tumori che sono caratterizzati, come elemento distintivo, da alterazioni genetiche peculiari. La più frequente è associata a traslocazione del gene TFE3: si tratta di riarrangiamenti che riguardano un gene (porzione di un cromosoma) che si trova sul cromosoma X e che portano alla crescita e alla moltiplicazione cellulare incontrollata.
Per la diagnosi l’anatomopatologo, oltre alle colorazioni dei vetrini di routine, si dovrà avvalere della metodica molecolare in situ FISH (fluorescent in situ hibridization), che consente la visualizzazione al microscopio di particolari alterazioni dei geni, attraverso l’utilizzo di sonde fluorescenti che legano specifiche aree del DNA.
Carcinoma associati a carenze di enzimi (succinato deidrogenasi e fumarati idratasi)
Carcinomi renali, rari, legati a mutazioni germinali (presenti dalla nascita) nei geni legati alla costituzione di un enzima, la succinato-deidrogenasi.
Hanno una prevalenza maschile (2:1) e l’età media di presentazione è intorno ai 40 anni.
Microscopicamente si tratta di tumori costituiti da cellule di forma cuboidale con citoplasma tipicamente di colora rosa (eosinofilo).
Per arrivare alla diagnosi l’anatomopatologo avrà necessità di richiedere ulteriori preparazioni, con l’ausilio di anticorpi specifici per l’enzima: questa reazione, data l’assenza dell’enzima risulterà negativa.
Vi sono altri tipi istologici, che si presentano con frequenza molto bassa, quali carcinoma midollare, tumori mesenchimali e carcinoma dei dotti collettori.
Tumori del rene in contesto ereditario
Esiste una quota di tumori del rene che insorge nel contesto di sindromi ereditarie. Tali tumori rappresentano circa il 2-4% delle neoplasie renali.
Di solito la presenza di un’alterazione genetica ereditaria si può sospettare in base all’età di insorgenza precoce, in base alla presenza di neoplasia renale multifocale e/o bilaterale, in base alla storia familiare del paziente o alla presenza di altre lesioni tipiche che caratterizzano le sindromi ereditarie.
Ci sono delle forme ereditarie e familiari che predispongono allo sviluppo di patologie neoplastiche benigne e maligne, tra cui carcinomi renali. Sono molto rare e la più importante è quella legata al gene VHL, la sindrome di von Hippel-Lindau, malattia genetica caratterizzata dalla predisposizione all’insorgenza di alcuni tipi di tumori, benigni o maligni in vasi organi ad esempio del sistema nervoso, emangioblastomi del cervelletto o del midollo spinale e angiomi retinici, o in vari organi, formazione di cisti o tumori endocrini (feocromocitomi). In questa sindrome si assiste ad un aumento della probabilità, sempre maggiore all’aumentare dell’età dei pazienti, di sviluppare carcinomi renali a cellule chiare.
Altra sindrome è la sindrome di Birt-Hogg-Dubè, molto rara, causata da una mutazione ereditata del gene della follicolina, che predispone allo sviluppo di: tumori del rene, che compaiono in media intorno ai 50 anni, neoplasie cutanee in corrispondenza dei follicoli piliferi e cisti polmonari, che possono rompersi causando pneumotorace.
Altre sindromi ereditarie sono la sclerosi tuberosa (che porta alla formazione di amartomi, cioè la crescita di tessuti non neoplastici in sede diverse del corpo), la sindrome del carcinoma papillare ereditario e la sindrome leiomiomatosi ereditaria – carcinoma renale (HLRCC), con mutazione germinale del gene della fumarato idratasi.
Grado istologico
Nel referto istopatologico, una delle caratteristiche importanti che viene riportata, è il grado. Quest’ultimo rappresenta lo stato di differenziazione della neoplasia ovvero ‘’quanto le cellule che la compongono si discostano da quelle del tessuto sano da cui hanno origine’’ ed è un indice sintetico che sta a rappresentare l’aggressività di una neoplasia.
Nel caso del tumore del rene viene studiato e riportato il grado dei tumori a cellule renali, cellule chiare e papillare, non nel cromofobo.
Le classificazioni utilizzate sono la classificazione di Fuhrman e quella del Sistema di classificazione OMS/ISUP. Entrambe si articolano su una scala che consta di 4 livelli di gravità crescente e il patologo prende in considerazione le alterazioni della cellula tumorale per inserire il tumore nella categoria di riferimento.
Un tumore ben differenziato, composto da cellule con alterazioni di forma, contorno, colore e dimensioni, lievi rispetto alle cellule non tumorali da cui originano, avrà un grado istologico G1.
Un tumore con una variabilità di forma e dimensioni marcata, cellule giganti con più di un nucleo (definite multinucleate) o cellule con caratteristiche dei nuclei particolari, dimensioni aumentate eccessivamente o che assumono un aspetto allungato o con un contorno particolarmente irregolare rispetto al comune aspetto tondeggiante (aspetti rabdoidi o sarcomatoidi) avrà un grado istologico G4, con prognosi molto peggiore e comportamento biologico più aggressivo.
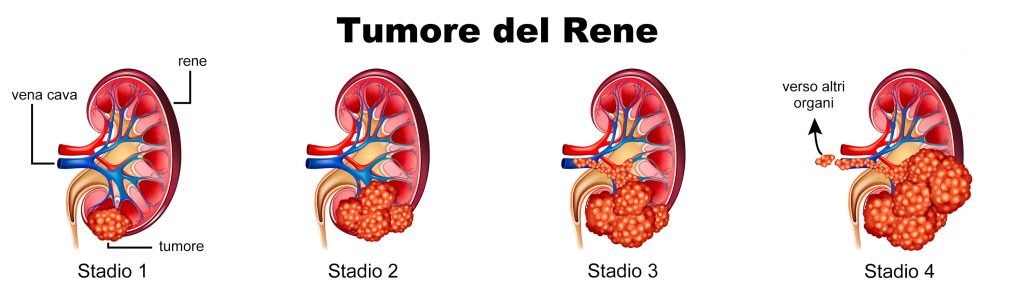
Stadiazione del tumore del rene
Un compito importante dell’anatomopatologo è quello relativo alla stadiazione del tumore. Lo stadio di un tumore ne descrive l’estensione, in termini di grandezza e/o coinvolgilmento di organi e tessuti adiacenti o a distanza.
Ad ogni stadio corrisponde una prognosi e a seconda dello stadio si ha accesso a diverse opzioni terapeutiche.
A tal fine si utilizza la classificazione internazionale del sistema TNM (acronimo inglese di Tumor, Node e Metastasis)
Per descrivere il tumore del rene il T, relativo alle caratteristiche del tumore, è accompagnato da un numero da 1 a 4.
- pT1 – per i tumori fino a 7 cm,
- pT2 – per i tumori fino a 10 cm,
- pT3 – per i tumori che si estendono alla vena renale e/o ai tessuti connettivi perirenali ma non oltre la fascia di Gerota, o alla vena cava inferiore,
- pT4 – per i tumori con infiltrazione oltre la fascia di Gerota (con possibile coinvolgimento di altri organi).
Per quanti riguarda la valutazione dei linfonodi, si avrà lo stadio:
- N0 in assenza di metastasi linfonodali;
- N1 se la malattia si localizza in almeno un linfonodo;
- Se non saranno analizzati linfonodi la sigla utilizzata sarà NX.
M0 è la sigla utilizzata in assenza di metastasi a distanza, M1 se presenti.

dr.ssa Caterina Marchiò
Dirigente Medico Anatomopatologo e Responsabile della Diagnostica Molecolare e delle Attività di Ricerca in Anatomia Patologica presso l’IRCCS di Candiolo

Dr. Leonardo Tonelli
Dirigente Medico Anatomopatologo presso l’IRCCS Istituto di Candiolo.